1. Introduction
Diabetic retinopathy (DR) is a common complication of diabetes. Elevated blood glucose levels induce alterations in a number of metabolic pathways that trigger microvascular lesions. It can cause significant vision deterioration due to development of macular edema or proliferative DR leading to intravitreal hemorrhages and tractional retinal detachment. Elaboration of effective methods in the treatment of DR is based on understanding of pathogenesis of this disease.
2. Blood flow changes
A hallmark of diabetes is a high blood glucose levels. It was shown in nondiabetic animals that infusion of glucose causes a rapid increase of retinal blood flow [1]. Patients with mild or no DR demonstrated significantly increased retinal blood volume flow compared with nondiabetic participants [2, 3]. Apparently, blood flow abnormalities contribute to the pathogenesis of DR and precede the earliest visible signs of diabetic retinal complications. Blood flow in the retina is controlled mostly by metabolic autoregulation.
Metabolic autoregulation is an adaptation of the diameter of vessels to the metabolic demands in the tissues. Oxygen saturation is a principal metabolic stimulus for blood flow changes in the retina. Impaired metabolic autoregulation in patients with DR may be due to changes in the retinal metabolism. It was founded that glucose flux through the polyol pathway in DR disrupts balance between pyruvate and lactate levels that resulted in pseudohypoxia and increased blood flow [4].
Probably, this is a direct mechanism of glucose-induced hyperperfusion in DR [5]. It was shown that increased blood flow induces increase in shear stress followed by a damage of the endothelial cell lining and basement membrane thickening [6]. Production of the vasoconstrictor endothelin-1 (ET-1) by endothelial cells is changed depending on the level and duration of the shear stress [7]. At the same time increased shear stress stimulates production of vasodilators, namely prostacyclin (PGI2) and nitric oxide (NO) followed by additional increase in blood flow [8, 9]. Hyperglycemia by its direct deleterious effect on pericytes inhibits contraction of small vessels, impairing autoregulation [10].
The dilatation of retinal vessels during the early stages of DR is accompanied with impaired pressure autoregulation. Pressure autoregulation is a capacity of the resistance vessels to adjust diameter to maintain stable microcirculation during changes in the arterial blood pressure. Thereby, in DR the systemic blood pressure more easily conveys to the capillary bed, increasing tangential stress on the capillary wall where it contributes to the formation of microaneurysms, hemorrhages, and breakdown of the blood-retina barrier (BRB) [6, 11].
The fact that hyperperfusion is essential in the development of DR is confirmed by conditions that are associated with its progression and characterized by increased blood flow. Hypertension is an important risk factor for the incidence and progression of DR [12, 13, 14]. Pregnancy in patients with diabetes is often associated with a deterioration in DR [15].
3. Microvascular changes
DR initially is a disorder of retinal capillaries that later propagates to the larger vessels. In the early stages of DR, microvascular lesions are characterized by development of microaneurysms, capillary leakage resulting in intraretinal hemorrhages, hard exudates, retinal edema, and also capillary occlusion resulting in ischemia and cotton wool spots formation.
More advanced stages of DR are associated with vascular changes such as vein beading, loop formation, intraretinal microvascular abnormalities (IRMA). DR progression leads to neovascularization, intravitreal hemorrhages, expanding of fibrous tissue, causing retinal traction and detachment. Exudative or ischemic forms of the sight threatening diabetic maculopathy may develop in any stage of DR [16].
Long before any clinically visible alterations occur, histological and pathophysiological changes in the wall of the vessels develop, involving thickening of the basement membrane, loss of pericytes, disturbance of endothelial cell functions. A crucial role in the progress of the disease is played by pericytes, developmentally originating from mesoderm. Pericytes are located along the endothelial cell tube, embracing with their cytoplasmic processes endothelial cells and providing mechanical support for the capillary wall [17, 18].
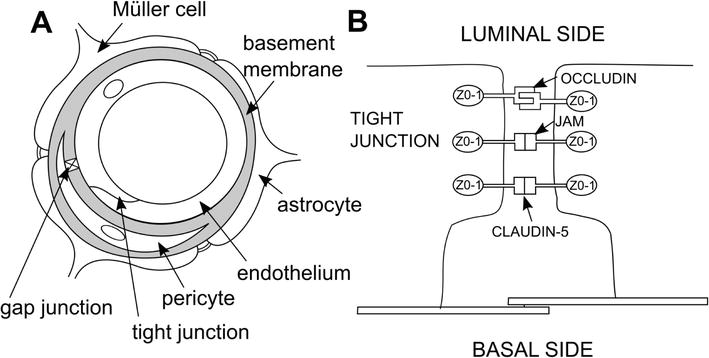
Pericytes are known as specialized contractile cells and function in the capillaries such as smooth muscle cells in the larger vessels, controlling vascular tone, and perfusion pressure [18, 19]. Pericytes are encased in a basement membrane (BM) that is continued with the endothelial BM (Figure 1A). The pericyte-endothelial cell interface is mainly divided by the BM. However, it was demonstrated that pericyte and endothelial cell plasma membranes contact across the BM fenestra [20].
There are different types of contacts described between endothelial cells and pericytes: peg-and-socket junctions, adhesion plaques, and gap junctions. In peg-and-socket contacts, cytoplasmic fingers of the pericytes interposed into the deep endothelial cell invaginations and, as assumed, support anchorage [21]. Adhesion plaques at the pericyte and endothelial cell plasma membrane serve as a mechanical binding among two cells, which allows the contraction or relaxation of the pericyte to be conveyed to the endothelial cell and thereby to affect capillary diameter [22].
Gap junctions are supposed to permit a direct connections between the cytoplasm of pericyte and endothelial cell [23]. It was proposed that ionic currents, the passage of small molecules and nucleotides, occur between endothelial cells and pericytes through the gap junctions [23, 24]. Moreover, it was shown that pericytes suppress capillary endothelial cell proliferation when cells are co-cultured in physical contact with each other, probably via gap junctions [25]. Interactions between endothelium and pericytes are also regulated by cell adhesion molecules, produced by both cell types, imbalance of which may cause leakage of the BRB during the early stages of DR [26].
Thereby, pericytes in the capillaries are closely associated with endothelial cells and regulate each other functions. Total cytoplasmic areas of the pericytes enveloping capillary and the cytoplasmic areas of the endothelial cells covering these capillaries comprise an average 1:1 ratio in human, which is much higher than that in other tissues [27, 28]. The cause for this high ratio is the necessity for an exceedingly high barrier function in the retina itself in order to prevent an extra fluid accumulation that could result in vision impairment. It seems that pericyte coverage in capillaries correlates positively with endothelial barrier characteristics in different tissues, and greater pericytes density apparently provides better integrity for the vasculature [27].
The BRB comprises the inner BRB (iBRB) and the outer BRB (oBRB). The iBRB is formed by the continuous lining of endothelial cells, tight junctions (zonula occludens) between adjacent endothelial cells and interconnecting pericytes. Tight junction proteins between apical regions of retinal pigment epithelial cells are structural components of the oBRB [29, 30]. The tight junctions are composed of integral membrane proteins, namely: claudin, occludin, junction adhesion molecules, and a number of accessory proteins such as zonula occludens −1 (ZO-1), ZO-2, ZO-3 (Figure 1B) [29, 31].
Pericytes are supposed to maintain the integrity of the iBRB by induction of expression of occludin and other junction proteins [30]. The early feature of DR is loss of pericytes, induced by high glucose levels that has been shown in a row of experiments. Naruse and colleagues demonstrated that high concentration of glucose inhibited proliferation of retinal capillary pericytes in the culture [32]. In particular, fluctuating glucose levels increased pericyte apoptosis in vitro [33].
Since pericytes are important compound of the capillary wall and maintain a capillary structure, loss of them results in localized outpouching of the microvessel wall. This process is linked with microaneurysms development, which is the earliest clinical sign of DR. Progressive pericyte apoptosis in complex with hypoxia causes dilation of the capillaries, venous caliber abnormalities such as venous beading and venous loops. Microaneurysms and dilated capillaries are usually incompetent and leaky [16]. Pericyte loss is accompanied by dysfunction and apoptosis of endothelial cells as well. Endothelial cells play an important role in the regulation of capillary permeability and tone.
These cells are responsible for metabolism of BM, coagulation balance, migration, and adhesion of leucocytes to the vessel wall, production of ET-1 [34]. It was demonstrated in vitro that endothelium in high glucose conditions secreted more BM material such as collagen and fibronectin IV, and overexpression of these products remained detectable even after endothelial cells were returned to normal glucose exposure [35, 36]. Thickening of BM in the early phase of DR may prevent endothelium-pericytes contacts that increases pericyte apoptosis due to deprivation of nourishment, while endothelium, losing control of proliferation from pericyte is involved in the formation of new vessels in later stages of retinopathy. Thickened BM reduces a diameter of affected vessels and facilitates capillary obliteration.
Dolgov and colleagues demonstrated weakening of endothelial intercellular gap junctions in the vessels during DR [37]. It was shown an increased apoptosis in cultured endothelial cells exposed by high glucose levels [38]. Furthermore, high glucose affects functions of endothelium indirectly by increased production of vasoactive agents and growth factors in other cells [39]. Thereby, DR progression leads to pericyte and endothelium pronounced disappearance, thickening of BM, and formation of acellular capillaries (tubes formed by basement membrane only), capillary occlusion, and ischemia.
Non-perfusion in some capillaries induces hypoxia, dilatation, and increased intracapillary pressure in others. Thereby, loss of pericytes impaired functions and later apoptosis of endothelium resulted in progressive retinal ischemia and BRB breakdown. BRB disintegration may occur at the level of both the iBRB and oBRB, causing accumulation of intraretinal fluid and plasma proteins first of all in the inner and outer plexiform layers of the retina, which is visible ophthalmoscopically as intraretinal hemorrhages, retinal edema, and hard exudates.
Fluid accumulation in the macular region can cause a macular edema leading to neuronal distortion and visual impairment [16]. Diabetic macular edema may be focal or diffuse. Focal edema is mainly caused by leakage from microaneurysms, whereas diffuse macular edema is a result of generalized leakage from dilated capillaries throughout the posterior pole, which is coupled with occlusion of capillaries. Diabetic maculopathy can associate with ischemia as well, due to mostly capillary obliteration, which is the main cause of visual impairment in this case. Progressive vessel occlusion increases retinal hypoxia and leads to the formation of significant non-perfusion areas in the retina, cotton wool spots, or soft exudates and intraretinal microvascular abnormalities (IRMA).
Cotton wool spots develop in cases of retinal arteriole occlusion and focal ischemia, which courses blockage of axoplasmic current and accumulation of large spheroidal axon swellings (“cystoid bodies”) and intra-axonal organelles in the retinal nerve-fiber layer [40]. IRMA is a tortuous collateral vessel located midway between arteries and veins. It is hypothesized that IRMAs are either dilated preexisting capillaries or newly formed vessels developing due to obliteration of capillaries and ischemia. IRMAs practically have no leakage and usually do not cross major large vessels [41].
In response to tissue hypoxia, vascular endothelial growth factor (VEGF) is released and stimulates angiogenesis. New vessels usually emerge from venous part of the retinal vasculature and grow uncontrolled [42]. If these vessels break the inner limiting membrane, they are defined as retinal neovascularization. New vessels penetrate through the inner limiting membrane, proliferate along the posterior hyaloid. They are fragile and may tear leaking blood into the retina and vitreous.
Subsequently, a fibrovascular scar tissue grows from the retinal surface into the vitreous cavity. Fibrous tissue retraction may course tractional retinal detachment and vision loss [43, 44].
4. Metabolic pathways implicated in hyperglycemia-induced lesion of vasculature
The Diabetes Control and Complications Trial (DCCT) clinical trial confirmed that chronic hyperglycemia is detrimental in the development and progression of DR, though the exact mechanisms of microvascular lesions due to hyperglycemia are not yet fully understood [45].
A lot of interconnecting biochemical pathways are involved in hyperglycemia-induced vascular pathologies. Four major mechanisms explaining how hyperglycemia causes diabetic complications include: (1) increased glucose flux through the polyol pathway, (2) increased formation of advanced glycation end products (AGE), (3) activation of the protein kinase C (PKC) pathway, and (4) a fourth mechanism has been suggested recently: increased glucose metabolism through the hexosamine pathway [46].
4.1 Increased polyol pathway flux
Excessive glucose in diabetes is metabolized through the polyol pathway. In the polyol pathway, aldose reductase (AR) reduces glucose to sorbitol (polyol), using nicotinamide adenine dinucleotide phosphate (NADPH) as a cofactor. Subsequently, sorbitol is slowly metabolized into fructose by sorbitol dehydrogenase (SDH) with NAD+ reduced to NADH.
Sorbitol is a sugar alcohol and strongly hydrophilic; therefore sorbitol cannot diffuse easily across the cell membrane. It was demonstrated that excessive intracellular storage of sorbitol results in hyperosmotic cellular damage [47]. Increased polyol pathway flux is considered to have several negative effects on retinal cells. Concomitant decrease of NADPH results in less NADPH availability for use by glutathione reductase, which uses NADPH as a cofactor to regenerate intracellular glutathione. Glutathione protects cells by neutralizing reactive oxygen species (ROS).
Thereby, reduced NADPH in a hyperglycemic environment could course or exacerbate intracellular oxidative stress. Fructose produced by the polyol pathway is metabolized consequently to fructose-3-phosphate or 3-deoxyglucosone, which are potent glycating substances and can lead to generation of AGEs [48]. The presence of AR was shown in the ganglion retinal cells, Müller cell processes, retinal pigment epithelium, and the pericytes and endothelial cells of retinal capillaries in diabetic models in animals. These studies also pointed out increased apoptosis of pericytes due to AR activity [49, 50].
It was demonstrated in other work, however, the presence of AR in the cytoplasm of pericytes but not in endothelial cells in experimental diabetes [51]. Sato and colleagues observed accumulation of sugar alcohols in pericytes in contrast with similar cultured endothelial cells [52]. It was shown that AR was localized in human retinal pericytes but not in the endothelial cells [53, 54]. This data suggested that a selective degeneration and loss of retinal pericytes may be due to AR activity. Some studies showed that aldose reductase inhibitors (ARIs) were able to reduce the incidence and severity of diabetic retinal lesions occurring in the galactose-fed animals.
The administration of ARIs to animal model of diabetes indicated that ARIs can prevent pericyte loss, formation of microaneurysms, hemorrhages, and abnormal growth of endothelial cells in areas of pericytes loss [55, 56]. Thickening of basement membrane in the retinal capillaries was significantly inhibited by administration of ARI in animal diabetic model [57]. It was shown in human genetic studies that certain polymorphisms of the AR gene are associated with elevated tissue levels of AR and higher risk of diabetic complications [58]. However, sorbinil retinopathy clinical trial, where ARI sorbinil was administered for 2–3 years to adults with insulin-dependent diabetes, had no clinically important effect on DR [59]. Probably, it is important to develop more effective ARIs.
4.2 Increased formation of advanced glycation end products (AGE)
AGEs are built up at a permanent but slow rate starting at the embryonic development, accumulate through entire life, and linked with aging. However, AGEs formation is markedly accelerated in diabetes because of hyperglycemic environment [60]. AGEs are formed from the non-enzymatic reaction of sugars, such as glucose and fructose with free amino groups of proteins, lipids, and nucleic acids.
The initial products of this reaction, such as Schiff bases, which spontaneously reform themselves into Amadori products, are reversible. Further reactions and molecular rearrangements result in the formation of irreversible crossed-linked derivatives termed AGEs, which are composed of a heterogeneous class of molecules that are yellow brown pigments, fluoresce. AGEs capable of forming cross-links with other structures and interact with cells via specific cell-surface AGE-binding receptors (RAGE), triggering inflammatory events, production of growth factors, generation of reactive oxygen intermediates induce oxidative stress [61, 62].
AGEs are toxic because they can modify intracellular proteins, including those involved in the regulation of gene transcription, or transfigure the extracellular matrix proteins, leading to reduction of the cell-to-cell interaction and vascular dysfunction, and also can modify circulating blood proteins. It demonstrated free radical generation by glycation products in vitro [63, 64]. The interaction of AGEs with RAGE has been involved in the development of DR.
It was demonstrated that retinal endothelial cells, pericytes, and ganglion cells are expressed RAGE under normal or diabetic conditions in vitro and in vivo [61, 62, 65, 66]. Yatamagishi and colleagues demonstrated pericytes apoptosis mediated via AGEs-RAGE interactions. It was proposed that AGEs-RAGE interactions induced generation of intracellular ROSs, which course overexpression of proapoptotic Bax protein in pericytes [67]. Schmidt studies demonstrated that AGEs after interaction with their cellular receptors are responsible for induction of oxidative stress, activation of nuclear factor kappa-light-chain-enhancer (NF- kB) both in vitro and in vivo [61, 62]. NF-kB is associated with transcriptional activation of genes associated with inflammatory responses [68].
Interestingly, persistent hyperglycemia leads to a gradual accumulation of AGEs in the BM and in pericytes in diabetic animal models, however, the retinal endothelial cells did not store AGEs. It was suggested that endothelium is capable to uptake AGEs directly from the blood stream through RAGE located on their luminal surface and further transfer the AGEs to subendothelial matrix and to pericytes. The preferential appearance of intracellular AGE deposits within pericytes and BM may affect their functions and lead to progression of DR [65].
Another study by Yamagishi demonstrated that in vitro exposure of retinal pericytes to AGEs retarded pericytes growth and induced apoptosis; moreover, these effects were cell-specific [66]. It was found that administration of an inhibitor of AGEs to diabetic animals prevented accumulation of AGEs in the retinal capillaries and significantly diminished pericyte loss, subsequent formation of microaneurysms, acellular capillaries, and capillary closure [69]. Furthermore, it was shown in vitro that AGEs induced VEGF overproduction by retinal pericytes, that is, additionally disturbed retinal microvascular homeostasis in concert with pericyte apoptosis [70]. Thereby, accumulation of AGEs significantly contributes to the development of diabetic retinopathy.
4.3 Activation of the protein kinase C (PKC) pathway
PKC activation has been shown to induce retinal vascular abnormalities in diabetes. Diacylglycerol (DAG) is the primary activator of PKC in physiology [4]. Increased total levels of DAG in DR were found [71]. Augmentation of DAG levels in diabetes can occur by several pathways. Hyperglycemia results in an increase of glucose flux through the glycolysis pathway, which in turn leads to enhanced de novo synthesis of DAG from glycolytic intermediates [72, 73, 74].
DAG can be gained as well from the hydrolysis of phosphatidylinositides, from the metabolism of phosphatidylcholine by phospholipase C [75]. Increased generation of DAG and the subsequent activation of PKC isoforms affect retinal functions in multiple different ways. Activation of DAG-PKC pathway is associated with cellular and vascular abnormalities in the retina such as increased endothelial permeability, basement membrane thickening, leucocyte adhesion, cytokine activation, abnormal angiogenesis, and excessive apoptosis [72].
Activation of PKC regulates gene expression via of phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathways. Induced by phosphorylation, in response to extracellular signals, MAPK and PI3K regulate functions of a broad array of proteins involved in cell growth, proliferation, motility, adhesion, survival, apoptosis, and angiogenesis [76, 77]. Among the various PKC isoforms, the beta-isoform seems to be activated preferentially in the vasculatures of diabetic animals [78]. It was demonstrated that PKC beta-isoform plays a role in the VEGF-induced vascular permeability in the retina of diabetic animals and VEGF-induced proliferation of endothelial cells. Furthermore, PKC beta-isoform-selective inhibitors decreased VEGF-induced vascular permeability and endothelial cell growth [77, 78].
VEGF is a dimeric glycoprotein and has a crucial role in the development and progression of DR. In mammals, the VEGF family comprises seven members where VEGF-A typically, and below, referred to as VEGF. VEGF regulates cell functions via vascular endothelial growth factor receptor-1 (VEGFR-1) and VEGFR-2, which belongs to the receptor tyrosine kinase family and primarily implicated in angiogenesis [79]. Hypoxia is the major inducer of increased VEGF transcription in the retina via hypoxia inducible factor-1 (HIF-1) [44, 80]. In addition to hypoxia, a number of other factors can stimulate the overexpression of VEGF in DR, including oxidative stress and insulin-like growth factor [81].
The pathways by which these factors regulate upregulation of retinal VEGF transcription are not yet understood. However, it has been demonstrated that ROS can induce VEGF transcription by a mechanism involving the activity of signal transducer and activator of transcription factor 3 (STAT3) [82]. It was found that increased vessel permeability is correlated with increased ocular levels of VEGF [83]. It was suggested that VEGF-induced permeability results from triggering of a cascade of proteolytic activities on the endothelial cell surface.
VEGF induces expression of urokinase plasminogen activator receptor (uPAR) that initiates cleavage of plasminogen by urokinase plasminogen activator (uPA). Subsequently, plasmin formation leads to activation of membrane-bound pro matrix metalloproteinase-9 pro (MMP-9) [84]. MMP-9 induces pericellular proteolysis affecting cell-cell and cell-BM attachment, producing leaky vessels and permitting to the endothelial cells to penetrate the underlying BM, migrate, and proliferate [85].
It was shown as well that VEGF increases microvascular permeability via increasing the intracellular calcium concentration in endothelial cells [86]. Growth of new blood vessels is induced by VEGF-VEGFRs mediated activation of MAPK cascades resulting in endothelium proliferation, migration, and tube formation [44]. Anti-vascular endothelial growth factor (anti-VEGF) drugs are viable treatment option for patients with diabetic macular edema and proliferative diabetic retinopathy [87, 88].
A role of PKC activation in the thickening of capillary basement membrane that is the prominent structural abnormality in the retinal microvessels in early DR was demonstrated. Treatment with PKC agonists stimulated type IV collagen expression and fibronectin accumulation that may increase the BM thickness [89, 90]. It was reported that inhibition of Na+ -K+ -ATPase by hyperglycemia was due to consecutive activation of PKC and cytosolic phospholipase A2 (cPLA2), inducing release of arachidonic acid and increased production of PGE2, which are known inhibitors of Na+,K(+)-ATPase [91]. Na+-K+-ATPase is a component of sodium pump, and it takes part in regulation of cellular contractility, MAPK transduction pathways, ROS formation, intracellular calcium levels.
PKC takes part in sustaining of chronic inflammation in DR. A row of studies were demonstrated that activation of PKC in endothelial cells triggered upregulation of intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule (VCAM)-1 that increased adhesion of leukocytes to the vascular endothelium. PKC inhibitors prevented upregulation of ICAM -I and (VCAM)-1 and adherence of neutrophils to endothelial cells [92, 93, 94, 95, 96]. It was demonstrated that leukocytes trapped in retinal vasculature course capillary occlusion, vascular cell, and BM alterations in the animal diabetic model [97]. Being adhered to the vessel wall, leukocytes may release ROS, enzymes, and cytokines, which damage the endothelial cells and increase vascular permeability [98, 99].
Taking in account the significance of pathological events inducing by activation of PKC, inhibitors of PKC have been studied as potential therapeutic agents for the treatment of patients with microvascular complications associated with diabetes [100].
4.4 Increased flux through the hexosamine pathway
The hexosamine biosynthesis pathway (HBP) is a comparatively minor part of glycolysis. Hyperglycemic condition increases glucose flux through HBP. High availability of intracellular glucose leads to an excess amount of fructose-6-phosphate. The largest proportion of fructose-6-phosphate is utilized in the glycolytic pathway. Glutamine: fructose-6-phosphate aminotransferase (GFAT) regulates the entry of fructose-6-phosphate into the HBP.
The major end product of the HBP is UDP-N-acetylglucosamine (UDP-GlcNAc) that catalyzes the addition of O-linked β-N-acetylglucosamine (O-GlcNAc) to serine and threonine residues of proteins [101]. O-GlcNAcylation is an important protein posttranslational modification (PTM) that involves the addition of O-GlcNAc moiety to the hydroxyl groups of serine and/or threonine residues of proteins.
Such as phosphorylation, protein O-GlcNAc modification can directly modify protein functions and also lead to the changes of gene expression [102]. Under conditions of sustained hyperglycemia that occur in diabetes, GFAT is upregulated, fructose-6-phosphate flux increases through the HBP and results in increase of O-GlcNAc-modified proteins. There are studies showing an association between elevated flux through HBP and insulin resistance [103]. It was demonstrated that high levels of O-GlcNAcylation of proteins in the retinal endothelial cells and pericytes correlated with glucose concentration levels, but the physiological consequences of this mainly remain unknown [104].
O-GlcNAc protein modification dysregulation under hyperglycemia and/or ischemia may contribute to the pathogenesis of the DR and retinal neovascularization [104, 105]. Decreasing glucose flux through the HBP by preventing the biosynthesis of UDP-GlcNAc would appear to reduce glucose toxicity, but would also induce adverse effects. A lot of proteins including kinases, phosphatases, transcriptional factors, and metabolic enzymes can be O-GlcNAc modified, but the functional consequences of this modification remain unknown for most of these proteins and need to be clarified.
5. Retinal neural and glial cell impairment
The neural retina is transparent and largely undistinguished during clinical examination in contrast to retinal vessels. Nevertheless, the retina comprises a complex network of neurons and glia closely interconnecting with vasculature.
The neurons: photoreceptors, bipolar cells, horizontal cells, amacrine cells, and retinal ganglion cells percept, integrate, and transmit visual signals into the brain. Glia comprises astrocytes, Müller cells (MCs), and microglial cells. MCs are the primary glial cells of the retina and play a pivotal role in retinal metabolism. It is now broadly acknowledged that in addition to the vascular alterations structural and functional detriment to nonvascular cells contributes to the pathogenesis of DR. Abnormalities of the neural retina have been found in experimental and human diabetes.
There is evidence demonstrating an early neurodegeneration of photoreceptors in animal diabetic models [106]. Apoptosis of retinal ganglion cells has been observed as well in cases of short-term experimental diabetes and in humans with diabetes [107]. Decline of color sensitivity [108] and contrast sensitivity [109] are early signs of neural retinal malfunction that take place after only 2 years of diabetes. As glia maintenance functions of neurons and endothelium, apparently, glial reactive changes affect the function and survival both of vascular and of neuronal cells of the retina.
It was detected that high glutamate levels in the retinas of diabetic animals as a consequence of MC reduced ability to convert glutamate into glutamine [110, 111]. Glutamate has been demonstrated to be toxic to neuronal cell [112, 113]. These findings suggested an early and probably persistent glutamate excitotoxicity in the retina during diabetes that courses neural degeneration. One of the early signs of retinal metabolic stress is the upregulation of glial fibrillary acidic protein (GFAP) in MCs and astocytes, which was detected in animals and in human patients with non-proliferative retinopathy [111, 114].
It was found that in activated MCs and astrocytes, VEGF expression was significantly increased [115, 116]. Glial cell proliferation is a well-recognized latest change in DR that induces epiretinal membrane formation, and fibrous tissue grows [43]. Microglia become activated early in diabetes in human and diabetic animal models [117]. It is supposed that diabetic conditions lead to an elevation of proinflammatory cytokine expression within the retina that induce microglia activation [118].
Being activated, microglia migrate to the source of inflammation and start to produce a wide range of proinflammatory cytokines, such as TNF- α, IL-6, IL-1, and IL-1, glutamate, ROS, NO, matrix metalloproteinases. All of these factors are implicated in the pathogenesis of DR, affect neuronal cell functions, and induce apoptosis [117, 118, 119, 120, 121]. It has been recognized that inflammation plays a pivotal role in pathophysiology of DR. Microglia, as highly sensitive to even low pathological changes in immune-effector cells in the retina, might be expected to have a significant role in the promotion and sustaining this inflammatory response.
6. Conclusion
Hyperglycemia induces a bewildering list of changes during DR in the retinal vasculature, neurons, and glia in animal models of diabetes and in diabetic patients. Apparently, increased flux of glucose and its metabolites affects a lot of cellular biochemical pathway driving a diverse nature of the changes.
The main challenge for research studies is to identify those hyperglycemia-induced biochemical alterations that are significant in causing vascular and neural pathologies for the development of new effective ways for the treatment of DR.
Author
Natalia Lobanovskaya
Department of Ophthalmology, East Viru Central Hospital, Kohtla-Järve, Estonia
Attribution
Lobanovskaya, Natalia. “Pathophysiology of Diabetic Retinopathy” In Diabetic Eye Disease: From Therapeutic Pipeline to the Real World, edited by Giuseppe Giudice. London: IntechOpen, 2022. 10.5772/intechopen.100588
